



Deep convolutional networks for quality assessment of protein folds
The computational prediction of a protein structure from its sequence generally relies on a method to assess the quality of protein models. Most assessment methods rank candidate models using heavily engineered structural features, defined as complex functions of the atomic coordinates. However, very few methods have attempted to learn these features directly from the data.
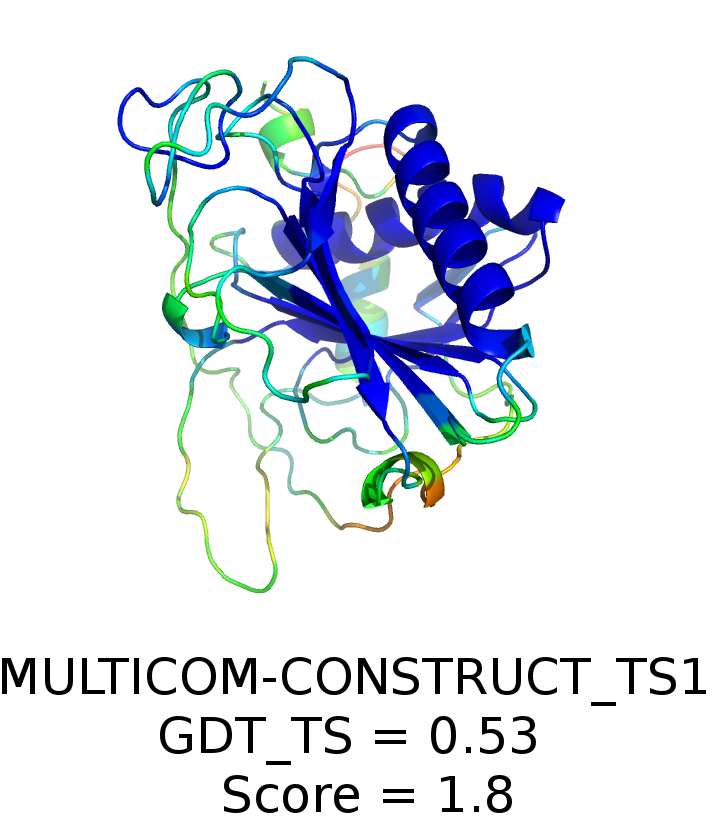
We show that deep convolutional networks can be used to predict the ranking of model structures solely on the basis of their raw three-dimensional atomic densities, without any feature tuning. We develop a deep neural network that performs on par with state-of-the-art algorithms from the literature. The network is trained on decoys from the CASP7 to CASP10 datasets and its performance is tested on the CASP11 dataset. Additional testing on decoys from the CASP12, CAMEO, and 3DRobot datasets confirms that the network performs consistently well across a variety of protein structures. While the network learns to assess structural decoys globally and does not rely on any predefined features, it can be analyzed to show that it implicitly identifies regions that deviate from the native structure.
The code and the datasets are available at https://github.com/lamoureux-lab/3DCNN_MQA.
Supplementary data are available at Bioinformatics online.
Reference
Georgy Derevyanko, Sergei Grudinin, Yoshua Bengio, Guillaume Lamoureux; Deep convolutional networks for quality assessment of protein folds, Bioinformatics, bty494
Source code
https://github.com/lamoureux-lab/3DCNN_MQA

